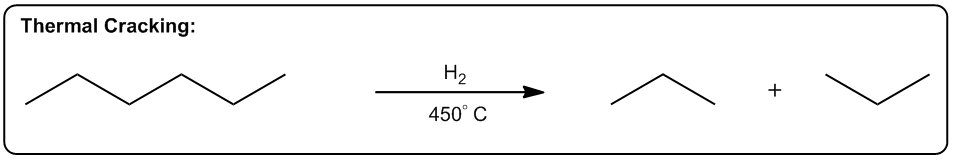
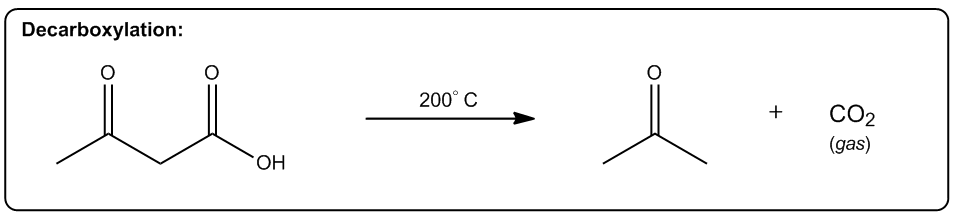
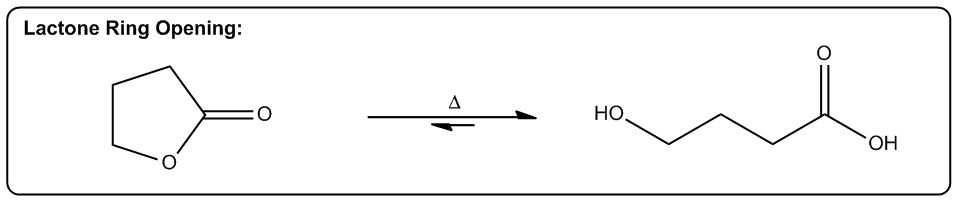
Entropy is a fundamental concept in thermodynamics, often described as a measure of disorder within a system. However, a more intuitive definition is that entropy reflects the tendency of a system to adopt its most probable state. In simpler terms, when comparing two states—one highly ordered and the other disordered—the disordered state is statistically more likely to occur. This probabilistic nature of entropy is crucial in understanding chemical reactions.
Even in highly exothermic reactions, where energy is released, the degree of order required can make such reactions statistically improbable. Therefore, analyzing whether a reaction will result in a positive or negative change in entropy (ΔS) is essential. A positive ΔS indicates an increase in disorder, while a negative ΔS signifies a transition towards greater order.
In practical terms, while calculating the exact value of entropy may not be feasible in introductory organic chemistry, students are expected to determine whether a reaction will lead to higher or lower entropy. This involves recognizing the conditions under which entropy increases, which are often favored by higher temperatures.
In summary, understanding entropy involves recognizing its connection to probability and disorder. The key takeaway is that reactions tend to favor states of higher entropy, particularly at elevated temperatures, which can influence the spontaneity of chemical processes.