All right, guys. So now I want to talk about what I would consider the most confusing term of the Gibbs Free Energy Equation, and that is entropy. All right. So entropy, generally stated, we said that it was a measure of disorder in the system. Okay? But that's a really confusing definition. I would rather go with an easier definition. Okay? And what that is, is that entropy is the tendency of a system to take its most probable form. Okay? So that means that if we have two different states, one is highly ordered and one is not as ordered, statistically, it's more likely to be in the disordered state. Okay? And that's what entropy has to do with. It has to do with probability really. Okay? So what that means is that even if a reaction is highly exothermic, like if you talk about bond dissociation energies and enthalpy, that has to do with exothermic. But the level of order that it requires might make it statistically improbable. Okay? So basically, what we do with entropy is we have to analyze whether this is going to be statistically more probable or statistically less probable. And that is what entropy is. Okay? Remember that we defined that entropy states that a negative value is going to be more ordered because remember that basically a positive value means that it's more disordered. That means that your entropy is getting bigger. So if you have a negative value, that means your entropy is getting smaller or it's getting more ordered. Okay? And it turns out that we're never going to have to calculate the entropy in terms of calculating the entropy of the surrounding environment or the entropy of a system because of the fact that we don't have really the tools to analyze that in orgo 1. Okay? But what you are going to be asked to do, okay, is you're going to be asked to analyze if something is going to have a positive ΔS or negative ΔS. And that's what we want to do. We don't want to figure out the exact number. We just want to figure out whether this is going to have higher entropy or lower entropy. Okay? Now, there is one situation where you might calculate ΔS, and that's if you're given every other variable. If you're given the T and the ΔH and the ΔG, then sure, you could just use algebra to figure out ΔS. But I'm just saying that in the absence of this being just a simple algebra problem, you're not going to be asked just to calculate what the ΔS of an environment. Okay? So let's talk about these three phenomena that make reactions more probable or make ΔS go up. And it turns out that all of them are going to be favored by high temperature. Okay? So that will be that should be really clear in a little bit when I go back to the equation.
Entropy - Online Tutor, Practice Problems & Exam Prep
 Created using AI
Created using AIEntropy is a measure of disorder, reflecting a system's tendency to adopt its most probable state. Reactions that increase the number of molecules, undergo phase transitions, or enhance molecular freedom of motion are entropically favored, especially at high temperatures. For example, thermal cracking breaks large hydrocarbons into smaller ones, increasing entropy. The Gibbs Free Energy equation, ΔG = ΔH - TΔS, shows that as temperature (T) rises, the significance of entropy (ΔS) in determining spontaneity increases, making reactions more favorable.
If a reaction is exothermic, shouldn’t that be enough to determine favorability? Actually, no!
Even if a reaction is highly exothermic, the level of order it requires may make it statistically improbable.
Explaining what entropy is.
Video transcript
Entropy (ΔS) is the tendency of a system to take its most probable form.
- Negative values (-) indicate less probable = Unfavored
- Positive values (+) indicate more probable = Favored
There are 3 common ways to make reactions more probable (increase ΔS)
- They all become more likely as we add heat to the reaction (increase T)
3 ways to increase entropy.
Video transcript
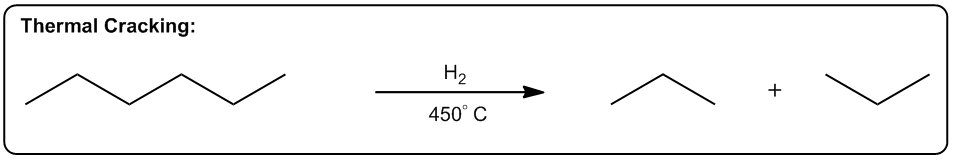
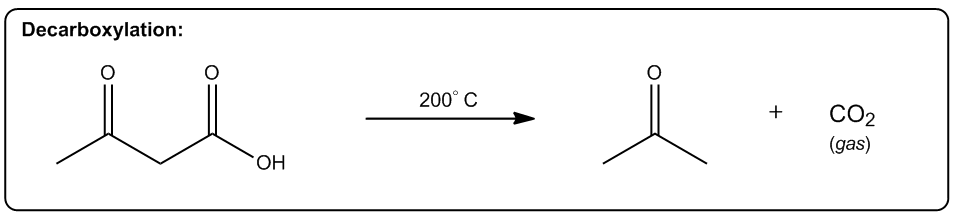
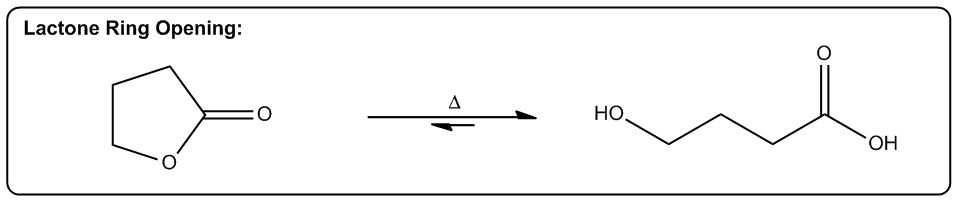
The first one is increasing the number of molecules in a reaction. Okay? So, any reaction that's going to make more moles of molecules or more equivalents of molecules after the reaction is over is going to be entropically favored. Why? Because what it does is it allows us to arrange these molecules in more ways. Alright? So here's a really good example, thermal cracking. Okay? This is used by the petroleum industry to take large hydrocarbons, ones that don’t really do anything. You can't put them in your car or anything. Then they put them with hydrogen in high heat. Okay? And what happens is that these long hydrocarbons spontaneously start breaking into smaller pieces. Okay? Why would this be a favored reaction? Okay? It actually takes energy to break those bonds, so why would these long hydrocarbons break? The reason is that at high enough temperature, what starts to happen is that if we can break 1 molecule into 10 molecules, right? That's going to be entropically favored because now I can arrange those molecules in different orders. Okay? So what that means is the chances are that if I have 2 systems that of equal scattered all over the place. Which one is going to be more probable? The one where it's scattered all over the place. Which one's going to be more probable? The one where it’s scattered. Okay? And that's why thermal cracking is favored. You take large hydrocarbons and you turn them into smaller ones by using high heat. High heat then makes the entropy part of the equation very, very favored. So now, the more pieces I can break it into, the more entropically favored that is. Alright? This is just one example, but there's lots of examples in organic chemistry of reactions that make more molecules than they start off with, meaning that they're entropically favored. Okay? So let's go on to the second one. The second one is a phase transition. Okay? If I can transform something from a solid to a liquid or from a liquid to a gas, that's going to be a more probable arrangement. The reason is because the molecules are now going to have greater vibrational freedom. Remember, the only difference between a solid, liquid, and gas is how much energy these molecules have. Okay? And how much motion they’re allowed to display. In a solid, there's very little vibration that can happen. They can only move in a very constrained space. K? Whereas in a gas, they're bouncing all over the place. For example, the gas in this room right now, it's bouncing off the walls, it's hitting me. That has a lot more freedom of motion than a solid does. So if I have 2 systems of equal energy, one where my molecules are constrained to one tiny little place next to another one, and then another system where the molecules can scatter as far as they want, which one is more favored? The one that they can scatter in. Okay? So what that means is that we have reactions that as you turn liquids into gases or solids into liquids, they're also entropically favored. A really good example is a reaction that we're going to learn in Orgo 2 called decarboxylation. Decarboxylation takes a carboxylic acid at high heat and it turns it into just a ketone and CO2 gas. Why is that favored? Because at high heat, now the entropy part of my equation becomes more powerful. It gets a higher weight. Okay? And if I’m turning something into a gas, that’s going to be more favored because a gas is more favored than a liquid. Okay? And by the way, this molecule right here would be a liquid at room temperature. Okay? Now, another thing to keep in mind is that this is also favored because of another thing, which is this is also favored by the making more molecules rule. Remember that here I have one molecule total. At the end of the equation, I have 1 molecule here and 1 molecule here, so I’m going from 1 mole to 2 moles of molecules. So what that means is that this is also going to be entropically favored not just because of the phase transition but also because I’m making a greater number of molecules. Alright? So let’s look at the last one. The last one is increasing, freedom of motion of a molecule. Okay? And this specifically has to do with cyclic molecules. If I can convert a cyclic molecule into an acyclic molecule, that's going to be more probable because now it's going to increase the freedom of rotation of these carbon chains. So this is also a reaction that you're going to see a lot in Orgo 2, lactone ring opening. Okay? A lactone is just a cyclic ester. Okay? That's the name of a lactone. We’re going to talk about that a lot more in orgo 2. What you see here is that in the presence of heat, what I can do is I can take my lactone and I can break the chain, the ring apart into 2 parts of the chain where this oxygen and this oxygen were linked together before and basically were the same thing. Okay? Now, what I do is I put 1 oxygen on each and now I have my carbon chain that has more freedom of motion. Now why is the chain better than the ring? Because remember when we talked about if remember when we talked about conformers and we talked about single bonds being able to rotate. Okay? What would happen is that these single bonds can all rotate a lot more than a ring. In a ring, you actually can't rotate at all. So what that means is that this one is going to have a whole lot more conformers possible than the first one. In fact, the first one doesn’t have any. Okay? That’s also more probable because if you have 2, molecules of equal energy and you have one where all of the bonds are eclipsed, that’s what this would be. This would be all eclipsed. Okay? If you have one where all the H's are eclipsed and then you have another one where they’re free to rotate and make anti and make gauche and make a bunch of different conformations. Which one's going to be more favored? The one on this side. So this one's also going to be entropically favored to open that ring as I increase the heat. As I jack up the heat more, that’s going to favor all of these reactions. And like I said, the reason has to do with Gibbs free energy. And remember that Gibbs free energy just basically stated that ΔG=ΔH-TΔS. So what that means is that as I increase T, what I'm going to do is I'm going to increase the amount that entropy matters. Okay? So if I raise all these reactions to a 1000 degrees, the enthalpy isn't even going to matter anymore. I'm not going to care about bond association energies because instead what I'm going to care about is how many molecules am I getting at the end or what's the freedom of rotation of these molecules. Interesting. That's going to tell us which one's more statistically favored and that’s going to be which one is more spontaneous. Alright?
1. Increasing the Number of Molecules
Reactions that create extra molecules are more probable since there are more ways to arrange them.
2. Phase Transition
Transformation of solid to liquid or liquid to gas is more probable since the molecules will have a greater vibrational freedom.
3. Increasing Molecular Freedom of Motion
Converting cyclic molecules to acyclic molecules are more probable since it increases freedom of rotation.
Do you want more practice?
More setsHere’s what students ask on this topic:
What is entropy in chemistry?
Entropy in chemistry is a measure of disorder or randomness in a system. It reflects the tendency of a system to adopt its most probable state. In simpler terms, entropy quantifies the number of ways a system can be arranged. Higher entropy means more disorder and more possible arrangements. For example, gases have higher entropy than liquids, and liquids have higher entropy than solids. The concept of entropy is crucial in understanding the spontaneity of chemical reactions, as described by the Gibbs Free Energy equation: .
Created using AIHow does temperature affect entropy?
Temperature significantly affects entropy. As temperature increases, the entropy of a system generally increases because the molecules have more kinetic energy and can occupy more possible states. This is particularly evident in phase transitions, such as when a solid melts into a liquid or a liquid evaporates into a gas. Higher temperatures make these transitions more likely, increasing the system's entropy. In the Gibbs Free Energy equation , a higher temperature (T) amplifies the impact of entropy (ΔS) on the spontaneity of a reaction.
Created using AIWhat is the relationship between entropy and Gibbs Free Energy?
The relationship between entropy and Gibbs Free Energy is described by the equation: . In this equation, ΔG represents the change in Gibbs Free Energy, ΔH is the change in enthalpy, T is the temperature in Kelvin, and ΔS is the change in entropy. A negative ΔG indicates a spontaneous reaction. As temperature increases, the term becomes more significant, meaning that reactions with positive ΔS (increased entropy) are more likely to be spontaneous at higher temperatures.
Created using AIWhy are phase transitions entropically favored?
Phase transitions are entropically favored because they increase the disorder and the number of possible arrangements of molecules. For example, when a solid melts into a liquid or a liquid evaporates into a gas, the molecules gain more freedom of motion and can occupy more states. This increase in molecular freedom and possible arrangements leads to higher entropy. In the context of the Gibbs Free Energy equation , the increase in entropy (ΔS) makes the phase transition more likely to be spontaneous, especially at higher temperatures.
Created using AIHow does increasing the number of molecules in a reaction affect entropy?
Increasing the number of molecules in a reaction generally increases the entropy of the system. This is because more molecules mean more possible ways to arrange them, leading to greater disorder. For example, in thermal cracking, large hydrocarbons are broken down into smaller molecules, increasing the total number of molecules. This increase in the number of molecules results in higher entropy, making the reaction entropically favored. In the Gibbs Free Energy equation , a positive ΔS (increase in entropy) contributes to a more negative ΔG, indicating a more spontaneous reaction.
Created using AIYour Organic Chemistry tutors


- For each reaction, estimate whether ΔS° for the reaction is positive, negative, or impossible to predict. a....
- When ethene is mixed with hydrogen in the presence of a platinum catalyst, hydrogen adds across the double bon...
- For each reaction, estimate whether ΔS° for the reaction is positive, negative, or impossible to predict. b....
- For each reaction, estimate whether ΔS° for the reaction is positive, negative, or impossible to predict. c....
- a. For which reaction in each set will ∆S° be more significant? b. For which reaction will ∆S° be positive? 1....
- a. Which of the following reactions has the larger ∆S° value? b. Is the ∆S° value positive or negative? a. b...
- At what temperature does the entropy change of a process not contribute to the favorability of a process?
- (••••) The hydrogenation of alkenes is a reaction we study in Chapter 9. (b) Is this reaction favored or dis...
- Through the course of this chapter, we have discussed only alkane chlorination and bromination, yet there are ...
- The dehydrogenation of butane to trans-but-2-ene has ΔH° = +116 kJ/mol (+27.6 kcal/mol) and ΔS° = +117J/kelvin...
- Estimate the value of entropy ( ∆S > 0 or ∆S<0 ) for the elimination step shown.
- Would you expect ∆S to be greater than, less than, or equal to zero in the following reactions? b.
- For each of the following processes, indicate whether you expect ∆S° to be greater than, less than, or equal t...
- For each of the following processes, indicate whether you expect ∆S° to be greater than, less than, or equal t...
- For each of the following processes, indicate whether you expect ∆S° to be greater than, less than, or equal t...
- Using the bond-dissociation energies in Table 5.6 (see Section 5.3.1), estimate the equilibrium constant of th...
- For the following reactions we have not seen yet, which side, if either, would be favored by increasing the te...
- Predict which side of the equilibrium is favored by ∆H, ∆G, and ∆S<IMAGE>
- For each of the following processes, indicate whether you expect ∆S° to be greater than, less than, or equal t...
- (••••) In Chapter 13, we discuss the ring-opening reactions of epoxides, such as the one shown here. <IMAGE...
- Would you expect ∆S to be greater than, less than, or equal to zero in the following reactions?a.
- For a reaction carried out at 25 °C with an equilibrium constant of 1 * 10-3, to increase the equilibrium cons...
- From the following rate constants, determined at five temperatures, calculate the experimental energy of activ...
- a. What is the equilibrium constant for a reaction that is carried out at 25 °C (298 K) with ∆H° = 20 kcal/mol...
- (ii) Which side of the reaction is favored by entropy? (iii) If ∆S° = 0 for these reactions, calculate ∆G° (...
- For the following values of ∆H° , ∆S°, and T, tell whether the process would be favored. (b) ∆H° = +7.34 kcal...
- Considering the process described in Assessment 5.13, will it be favored or disfavored at a temperature higher...
- For the following values of ∆H° , ∆S°, and T, tell whether the process would be favored.(d) ∆H° = -8.3 kcal/mo...
- Calculate ∆G°, ∆H°, and ∆S° for the following acid–base reactions. Rationalize the value of ∆H° based on the s...
- When ethene is treated in a calorimeter with H2 and a Pt catalyst, the heat of reaction is found to be -137 kJ...