Alright guys. So now we're pretty much pros at the SN2 mechanism, but it turns out that not all substitution reactions proceed through the SN2. There's actually another mechanism out there that also produces substitution at the end, and that mechanism is called SN1. Alright? So, if I were to just generally paraphrase what SN1 is, I would say the following sentence. Okay? And what you're going to notice is that quite a few of the keywords that we use for SN2 are going to change. So what it is, is that a neutral nucleophile, so that's a big deal, neutral, is going to react with an inaccessible leaving group. Okay? So what am I talking about here? Well, neutral means that, remember, SN2 is negative. So this one means neutral. It's not going to have that strong nucleophilic property. Okay? Inaccessible has to do with R groups. So, we're going to get there in a second, but you can already start to think that must mean that there are a lot of R groups maybe. Okay? And that's going to produce substitution in 2 steps. So, what you can see is that in this sentence, almost every word has changed except for substitution. What that means is, at the end, they still get a very similar product, which is a substitution product, but all of the conditions have pretty much changed. Now instead of being negative, it's neutral. Instead of being accessible, it's inaccessible. And instead of happening in one step, it's going to happen in 2. So let's just go ahead and start breaking this mechanism down. Okay? The biggest thing that you're going to notice that's different from about the SN2 versus the SN1 is that my nucleophile looks totally different. Before, my nucleophile was the initiator in the reaction. My nucleophile was like the arrow. It had a very strong negative charge and it was looking for any backside to react with. Okay? If I draw that same first step here, that's a huge mistake. Do you know why? Because this is not negatively charged, so it's not very nucleophilic. It's not looking for a backside right now. In fact, it's happy. It's neutral. So the nucleophile is just chilling. Right? So to just draw a coin in the backside would be a huge mistake. That nucleophile is chilling. It doesn't need to do anything. It's neutral. So that means that the first step must be something else. And it turns out that the first step is going to be very unusual, and I know that it's going to be weird, so I'm just going to try to get it over with. It's that the X or the halogen, the leaving group, is going to take off all on its own. Alright? So now, this is weird, because ever since I've been teaching you all about mechanisms, I've always said that you make a bond first and then after you break a bond. But here what I'm doing is I'm breaking a bond without making one first. I'm just breaking it by itself. Alright? Why is that possible? How does this even make sense? Well, unfortunately, I don't have another reaction in organic chemistry that I can compare this to. We've never done this before. But if you think way back when you took Gen Chem 2, we actually do know a reaction that was similar to this. I'll try to make the explanation quick because I know you barely remember Gen chem 2 probably. But remember when we were thinking back on acids and bases, and we learned about titrations, and we talked about how pH had to do with the amount of hydrogen ions you had and the OH ions. What we learned is that the K_w of water, the dissociation constant of water, was 1×10-14. What the hell does that mean? What it means is that water is normally H2O. Right? And that's the way it's happy. But for some random reason, one out of every 109 molecules is going to decide to just split apart on its own and make H+ and OH-. Okay? For a split second, it's going to do that. And guess what's going to happen? It's going to hate its life because now it's 2 ions and it's way less stable, and then it's going to come back together. Alright? This is a random process that's happening all the time. Okay? You can almost think of it like a divorce, but then they get right back together because they realize that they actually like each other. Okay? So in the same way, alkyl halides have a dissociation constant as well. So, I'm going to put here K_RX. But that dissociation constant is actually going to be a lot higher. Why? Because water is very unstable after it dissociates. But alkyl halides actually already have a really strong dipole. Now, on top of that, the bond, the carbon-halogen bond, is actually a very, very weak bond in comparison to the water. So what that means is that it's actually going to be easier for the alkyl halide to dissociate than water will be. So I don't know the exact number. I'm just going to make one up. Let's say that instead, it's 1×10-7. Okay? Now remember that this is on an exponential scale, so that doesn't mean it's twice as good. That means it's like a million times as good. All right? All I'm trying to say is that this is going to wind up giving me ions as well, but at a better rate than I would usually get for, or a better equilibrium than I would usually get for water. Okay? So what that means is I'm going to wind up getting C+ and I'm going to wind up getting X-. And that's what's happening here. All I'm trying to say is that random processes are going to drive my alkyl halide to ionize. And that's always going to be the first step. So what that means is that my nucleophile is chilling. It's neutral. But now, for some reason, my alkyl halide is going to ionize by itself. And what that's going to give me is a positive charge and a negative charge. The positive charge is what we're going to call our carbocation. Okay? Now, what our carbocation is going to look like is it's going to be this carbon right there. Okay, so I'm just going to draw that carbon. But now it's only attached to three things. It's attached to an H. It's attached to an ethyl group on one side. And it's attached to a methyl group on the other. Okay? Now, this carbon wants to have 4 bonds, but it only has 3, so it needs a positive charge. Okay? Now, do you remember what the hybridization and geometry was of a carbocation? Three bonds. It should be sp2. Okay. Because it only has 3 groups or 3 bond sites. And it should be trigonal
SN1 Reaction: Study with Video Lessons, Practice Problems & Examples
 Created using AI
Created using AIThe SN1 mechanism involves a neutral nucleophile reacting with an inaccessible leaving group, leading to substitution through two steps. The first step features the leaving group dissociating to form a carbocation, which is stabilized by surrounding R groups. The second step sees the nucleophile attacking the carbocation, resulting in a racemic mixture of products due to the planar structure of the intermediate. The reaction rate is determined by the slow formation of the carbocation, making it unimolecular. This process is often referred to as solvolysis, where the solvent acts as the nucleophile.
The SN1 mechanism is similar to SN2 in that you get a substitution product, but the path to get there is completely different. It’s important that we understand how it’s different from SN2.
Drawing the SN1 Mechanism
Video transcript
Summary: A neutral nucleophile reacts with an inaccessible leaving group to produce substitution in two-step.

Why highly substituted leaving groups favor SN1.
Video transcript
What I want to do is talk about the inaccessible leaving group part. Okay? And that has to do with R groups and it has to do with carbocations. So basically, there's this trend about carbocation stability that says that the more R groups that you have directly attached to a carbocation, the more stable it's going to be. Okay? And it turns out that since you need carbocations in order for the nucleophile to attack it, that means that the more R groups I have on my alkyl halide, the better it's going to be. So what that means is that a tertiary alkyl halide, or a tertiary, yeah, tertiary alkyl halide, is going to be way better at this than a primary. I'm just trying to point that out. Okay? Because a tertiary will be the one that's the most stable. And then this would be the side that's the most unstable. Okay? It should be unstable. I'm sorry. Not instable. Oh, my gosh. All right. Don't worry too much about this other stuff in the middle. I'm going to go through a much more robust explanation of carbocations in a little bit. Okay? But for right now, just know that the more R groups, the better. The fewer R groups, the worse. Okay?
The slow step of this mechanism is the formation of a carbocation intermediate. These types of intermediates are unstable, so anything that we can do to stabilize them will help them form faster.
-R groups stabilize carbocations through a phenomenon called hyperconjugation. Meaning that the more substituted the carbocation, the more stable it is.
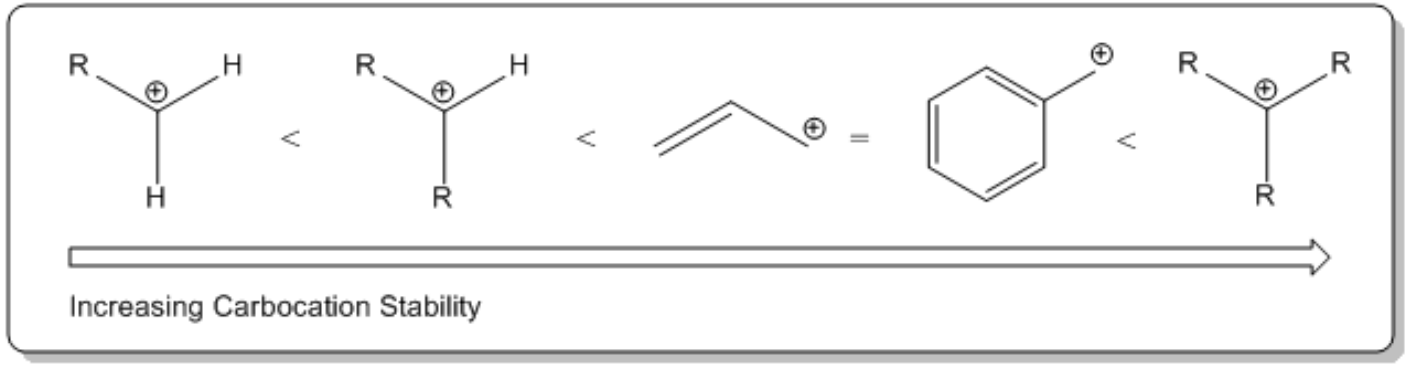
Understanding the properties of SN1.
Video transcript
So now let's go over our facts. Okay. As you can already see, this is going to be interesting. I've got an interesting explanation for you guys. So should the nucleophile be strong or should it be weak? It has to be weak. The reason is that if it were strong, it would do an SN2. It would do a backside attack. But since it's weak, it's not going to do anything. It's just going to chill and wait around for something to happen. Okay? Should my leaving group be unsubstituted, meaning no R groups or highly substituted, meaning lots of R groups? Highly, right? Because we just said that the more R groups you have, the more Rs, the more stable your carbocation is going to be and the carbocation is part of the mechanism, so we want more carbocations. Alright? Cool.
Is the transition in the reaction coordinate going to be a transition state or an intermediate? It's going to be an intermediate this time because I have distinct two different steps. Okay? So what that means is the thing in the middle, I could actually stop there if I wanted to and leave it there. But then later on, I probably don't want to keep it there, so that's why I'm going to go all the way to completion. That means that is it concerted? No. It's 2 steps.
Now I do want to show you guys that we actually have 2 different steps in terms of, well, you know what, I'm going to get to that in a second. Let's just keep going to the rate part and then I'm going to explain that. Okay? So now the rate. Should it be unimolecular or bimolecular? Well, it turns out that the name itself tells you the answer to this. Sn1, I'm not going to write out the whole thing, but it stands for substitution, nucleophilic, and then unimolecular. Okay? So that's what Sn1 means.
But how does that actually make sense? So let's go ahead and circle that. Okay? The only way to really know what that means is to think back at the way the mechanism works. And it turns out that in this mechanism, there is a slow step and there is a fast step. And the rates of your overall product are going to be determined by the rate of your slowest step, kind of like you're only as strong as your weakest link. It's a common phrase. The same thing, you're only as fast as your slowest step. Right?
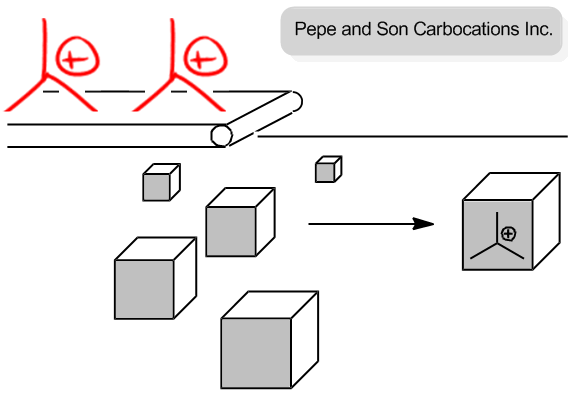
Let's pretend that we owned a theoretical company. We're going to go into business together. Alright? And we're Pepe and Sun carbocations. Alright? Our entire product is just to put carbocations inside of boxes and we sell people carbocations. We're going to make lots of money. Are you excited about this business? Cool. Me too.
So the coolest best thing about this business is that there are only 2 things that we need. The only things we need are carbocations, which come out on this conveyor belt. Okay? And then the other thing that we need is just boxes. Okay? So basically, I'll make the carbocations, you make the boxes, and then at the end, we'll sell these carbocations for a lot of money. Are you cool with that business strategy? You might want to change the name, but the business sounds pretty good. All right? So I'm going to make carbocations. You're going to make boxes.
Now, let's talk about ways that we could maybe get out more product and make more money. So how about if you make like a 100 more boxes than I make carbocations? So what if I go ahead and I'm making these carbocations one by one and you decide that you're going to just try to increase our sales and just make a 100 more boxes, is that going to increase the amount of products that we sell at the end? And the answer is no because this conveyor belt is going at a constant speed, and these carbocations are dropping into the boxes. Alright?
So even if I have a 100 more boxes here and I'm — this is wow. I'm really bad at drawing cubes. Okay? Even if I added a 100 more boxes here, would that change the amount of products that I get? No. Because I'm still waiting for the carbocations to drop one by one. Okay? So what that means is that the slow step or the rate-determining step here isn't how fast you can make a box. It's how fast the conveyor belt can drop a carbocation into the box. Alright?
Now, let's ask the other question. Okay? Let's say that I were to — and by the way, let's also pretend that there are a lot more boxes than there are carbocations. Okay? Because like I said, the boxes are easy to make. Okay? So what if I were to double the speed at which the conveyor belt drops carbocations? Would that increase the rate of my production or make more money? Yes, it would. Because again, we're assuming that the box is the easy thing to have, so we have tons of extra boxes, but the carbocations are the slow step. So if I were to just speed up the rate by 2, let's say now I have twice as many carbocations, now I'm going to make twice as many products because the box is the easy part. The hard part is making the carbocations. Are you guys getting that? That's the whole concept of the rate-determining step. And in this case, the rate-determining step is the carbocations being generated. Okay?
So what that means is that my nucleophile was the box. The weak nucleophile was the box. The leaving group is what makes the carbocation. Okay? So if I increase the concentration of the nucleophile, basically saying let's say that I double the amount of boxes that I have or double the amount of nucleophile, a weak nucleophile, is that going to increase my reaction? No. How would if I double the amount of leaving group? Yes. That's going to increase the rate because the leaving group is what turns into a carbocation. If I have twice as much leaving group, that's going to mean twice as much of a possibility that my leaving group randomly dissociates and makes a new carbocation. Is that kind of making sense? All right.
So what that means is that the rate's constant is going to be just the dissociation constant times the alkyl halide, not times the nucleophile because the nucleophile doesn't matter. Like I said, the nucleophile is just like the boxes. I have tons of extra boxes. If I increase the amount of boxes, that just means that I have boxes lying around. It doesn't mean that I actually make more product. Okay?
But if I increase the amount of my alkyl halide, that's like me speeding up the conveyor belt because now I'm going to make more carbocations than before. Cool. So that means that if we were to use this information on the top, what I would say is that the first step is my slow step. Okay? And then I would say that the second step or the attack is my fast step. The fast step is my carbocation getting attacked. That's easy to do. The hard part is making these carbocations. Okay? I hope that my analogy didn't confuse you guys more. I'm just trying to show you guys the difference.
Now, what's the stereochemistry? Is it going to be inversion, retention, or racemic? The answer is racemic. Okay? The reason why the answer is racemic is because like I said, this is trigonal planar. The intermediate is trigonal planar. So I have a 50% shot of hitting the front. And I've got a 50% shot of hitting the back. In this case, one of these is going to be R, one of them is going to be S. This would be R. This would be S. I have a racemic mixture, so I would just get even enantiomers of both. Okay? And that's also very important to remember about SN1. And the nickname for this, it's going to be a weird nickname, but it's called solvolysis. The reason is that our neutral nucleophiles are usually just going to be solvents. These are going to be our most common reagents that we use as nucleophiles. As you can tell, these are not very strong nucleophiles, but these are all typically solvents that wind up breaking. Solvolysis literally means that the solvent is breaking apart, and that's what winds up happening after the reaction. Okay?
You are a manager at Pepe and Son Carbocations Inc, and your boss has asked you to increase production of your product (carbocations in a box- these are awesome).
You have plenty of boxes, but it takes time for the conveyer belt to crank out these custom carbocations.
- Will increasing the number of boxes increase the amount of product?
- Will increasing the speed of the conveyer belt increase the amount of product?
Properties of SN1 reactions:
- Nucleophile = Weak
- Leaving Group = Highly Substituted
- Reaction coordinate = Intermediate
- Reaction = Two-Step
- Rate = Unimolecular
- Rate = k[RX]
- Stereochemistry = Racemic
- Nickname = Solvolysis
The product must be a racemate if the original leaving group is located on a chiral center.
NOTE:Substitution reactions with neutral nucleophiles require an additional deprotonation step.
Provide the mechanism and final products for the following reactions.
Predict the product of the following reaction:





Do you want more practice?
More setsHere’s what students ask on this topic:
What is the SN1 reaction mechanism?
The SN1 reaction mechanism involves two steps. First, the leaving group dissociates from the substrate, forming a carbocation intermediate. This step is slow and determines the reaction rate. The second step involves the nucleophile attacking the carbocation, leading to the formation of the substitution product. The intermediate carbocation is planar, allowing the nucleophile to attack from either side, resulting in a racemic mixture if the product is chiral. The reaction is unimolecular, meaning the rate depends only on the concentration of the substrate.
Created using AIWhy is the SN1 reaction rate unimolecular?
The SN1 reaction rate is unimolecular because it depends solely on the concentration of the substrate, not the nucleophile. The rate-determining step is the formation of the carbocation intermediate, which occurs when the leaving group dissociates from the substrate. This step is slow and determines the overall reaction rate. Since this step involves only the substrate, the reaction is classified as unimolecular.
Created using AIWhat factors affect the stability of the carbocation in an SN1 reaction?
The stability of the carbocation in an SN1 reaction is influenced by the number and type of R groups attached to the positively charged carbon. More R groups (alkyl groups) increase stability through hyperconjugation and inductive effects. Tertiary carbocations are more stable than secondary, which are more stable than primary. Additionally, resonance stabilization and the presence of electron-donating groups can further stabilize the carbocation.
Created using AIWhat is the stereochemistry of the SN1 reaction product?
The stereochemistry of the SN1 reaction product is typically racemic. This occurs because the carbocation intermediate is planar, allowing the nucleophile to attack from either the front or the back with equal probability. If the substrate is chiral, this results in a 50:50 mixture of enantiomers, leading to a racemic mixture.
Created using AIWhat is solvolysis in the context of SN1 reactions?
Solvolysis in the context of SN1 reactions refers to the process where the solvent acts as the nucleophile. Common solvents like water or alcohols can participate in the reaction, attacking the carbocation intermediate formed after the leaving group dissociates. This often leads to the formation of substitution products where the solvent molecule is incorporated into the final product.
Created using AIYour Organic Chemistry tutors


- Because the SN1 reaction goes through a flat carbocation, we might expect an optically active starting materia...
- Allylic halides have the structure H2C=CHC(X)H2 c. Show the products expected from SN1 solvolysis of these...
- Give the substitution products expected from solvolysis of each compound by heating in ethanol. (a) (b)
- A reluctant first-order substrate can be forced to ionize by adding some silver nitrate (one of the few solubl...
- Propose a mechanism involving a hydride shift or an alkyl shift for each solvolysis reaction. Explain how eac...
- Give the SN1 mechanism for the formation of 2-ethoxy-3-methylbutane, the unrearranged product in this reaction...
- Propose an SN1 mechanism for the solvolysis of 3-bromo-2,3-dimethylpentane in ethanol.
- Predict the compound in each pair that will undergo solvolysis (in aqueous ethanol) more rapidly. a. (CH3CH2)...
- Choose the member of each pair that will react faster by the SN1 mechanism. a. 1-bromopropane or 2-bromopropa...
- Furfuryl chloride can undergo substitution by both SN2 and SN1 mechanisms. Because it is a 1° alkyl halide, ...
- Optically active butan-2-ol racemizes in dilute acid. Propose a mechanism for this racemization.
- Draw the stereoisomers that are formed from the following SN1 reactions: a. 3-bromo-3-methylpentane and metha...
- Rank the following from most reactive to least reactive in an SN1 reaction:
- c. Which reacts faster in an SN1 reaction?
- (••••) The bromoalkanes shown below participate in Sₙ1 reactions with the relative rates shown. Explain this t...
- Practice your electron-pushing skills by drawing a mechanism for the following Sₙ1 reactions. (b)
- Practice your electron-pushing skills by drawing a mechanism for the following Sₙ1 reactions. (c)
- Provide a mechanism for the following Sₙ1 reactions that feature a rearrangement. (a)
- Which of the following Sₙ1 reactions should proceed at a faster rate? Justify your answer on a reaction coord...
- Practice your electron-pushing skills by drawing a mechanism for the following Sₙ1 reactions. (a)
- The allylic bromide below gives two Sₙ1 products. Justify the formation of each.<IMAGE>
- Rank the ability of the following compounds to undergo an S_N1 reaction ( 1 = fastest, 5 = slowest ).
- Propose a mechanism for each reaction, showing explicitly how the observed mixtures of products are formed. (b...
- Treatment of an alkyl halide with AgNO3 in alcohol often promotes ionization. Ag+ + R-Cl —> AgCl + R+ When...
- When 3-bromo-1-methylcyclohexene undergoes solvolysis in hot ethanol, two products are formed. Propose a mecha...
- Which haloalkane would you expect to undergo the fastest S_N 1 reaction? Why?
- Provide a mechanism for the following S_N1 reaction. What roles (acid/base/nucleophile/electrophile) does etha...
- (••) Predict the product(s) that would result when molecules (a)–(p) are allowed to react under the following ...
- (••) Predict the product(s) that would result when molecules (a)–(p) are allowed to react under the following ...
- Which compound is more reactive in an SN1 reaction? In each case, you can assume that both alkyl halides have ...
- a. Assuming that the two compounds shown below have the same stability, which one would you expect to be more ...
- A reluctant first-order substrate can be forced to ionize by adding some silver nitrate (one of the few solubl...
- Propose a mechanism involving a hydride shift or an alkyl shift for each solvolysis reaction. Explain how each...
- Choose the member of each pair that will react faster by the SN1 mechanism.c. n-propyl bromide or allyl bromid...
- Predict the compound in each pair that will undergo solvolysis (in aqueous ethanol) more rapidly.c. <IMAGE&...
- Allylic halides have the structureH2C=CHC(X)H2 <IMAGE> a. Show how the first-order ionization of an all...
- Often, compounds can be synthesized by more than one method. Show how this 3° alcohol can be made from thefoll...
- Give the substitution products expected from solvolysis of each compound by heating in ethanol.(c) <IMAGE&g...
- <IMAGE>Propose a mechanism for the reaction of benzyl bromide with ethanol to give benzyl ethyl ether (s...
- Propose a mechanism involving a hydride shift or an alkyl shift for each solvolysis reaction.Explain how each ...
- Predict the products of the following SN2 reactions.c. <IMAGE> Na+ + Ch3CH2Br —>d. CH3(CH2)8CH2Cl + N...
- Choose the member of each pair that will react faster by the SN1 mechanism.e. 2-iodo-2-methylbutane or tert-bu...
- When tert-butyl bromide is heated with an equal amount of ethanol in an inert solvent, one of the products is ...
- tert-Butyl chloride undergoes solvolysis in both acetic acid and formic acid. Solvolysis occurs 5000 times fas...
- Rank the following alkyl halides from most reactive to least reactive in an SN1 reaction: 2-bromo-2-methylpent...
- Which reaction would be faster, the one with DMSO as the solvent or the one with ethanol (EtOH)?
- a. Identify the substitution products that form when 2-bromo-2-methylpropane is dissolved in a mixture of 80% ...
- Explain why tetrahydrofuran can solvate a positively charged species better than diethyl ether can.
- Two bromoethers are obtained from the reaction of the following alkyl dihalide with methanol. Draw the structu...
- In contrast, optically active butan-2-ol does not racemize on treatment with a solution of KOH. Explain why a ...
- Propose a mechanism for each reaction, showing explicitly how the observed mixtures of products are formed. (e...
- There were actually two possible products in the solvolysis reaction from Figure 21.10. Show both products. Wh...