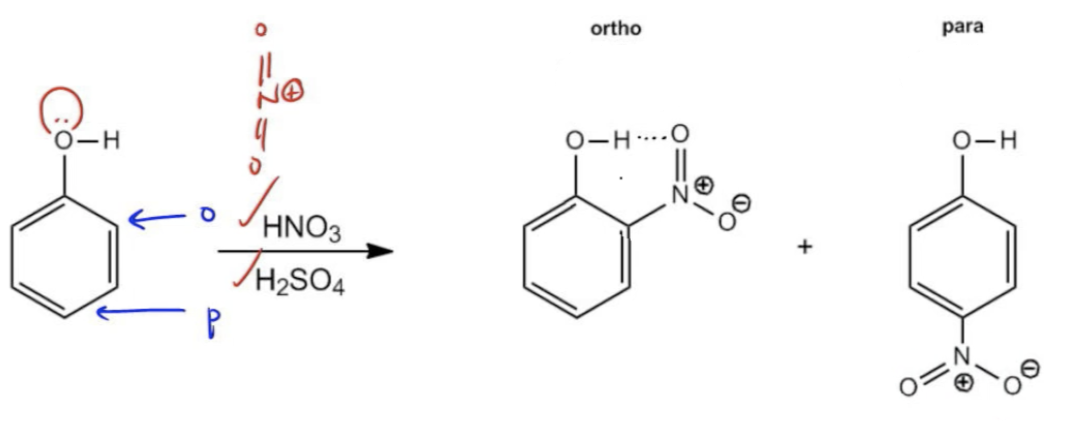
In this video, we're going to go into a little bit more depth on the major products of ortho para directed reactions. In general, whenever we have an EAS reaction on an ortho para director, we're just going to say that the product is a mixture of both the ortho and the para positions. If you want to play it safe, you can always just say that it's a mixture. And that's the way that most textbooks teach it. However, there are a few patterns that might be helpful for us to learn, especially later on when we start talking about specific groups called blocking groups. Let me tell you about the difference between the ortho position and the para position. Well, basically these positions are in competition with each other because they both have advantages. Let's say they have an electron donating group. That means it's pushing electrons into the ring and it's probably an ortho para director, right? Well, the ortho position has an advantage, a clear advantage right from the beginning because notice that it has two positions instead of one. The para position, there's only one para. But there are two ortho positions. So you might think, well we're going to get more ortho product because you have twice as many positions that could react. Okay? So that's one way to think about it. There are two times as many positions. But on the other hand, the para position also has an advantage which is that the para position is usually the one that's less hindered because it's furthest away from the donating group. So it's actually easier to add to that para position even though there's less of them. And in general, when you kind of pair these two together, when you compete them against each other in number versus steric hindrance, steric hindrance is usually going to win. So usually there's going to be a slight advantage for the para position over the ortho position. So if in a question you're asked to supply one major product instead of a mixture, if it says specifically which one is the major product, here we can see a sulfonation on toluene. You can see we've got our SO₃. We've got our fuming H₂SO₄ and look what we're going to get. What we are going to get is a major product of para. You would say that your para sulfonic acid is going to be the major product and your ortho is going to be the minor. Even though there are more ortho positions, the steric hindrance, the benefit of sterics is going to make the para the major product. Basically, that's going to be the rule that we always follow. We're going to say that para wins out over ortho if asked to supply one major product. Now there is one noted exception and that one exception is if the final product can hydrogen bond with itself. If the final product can hydrogen bond because of the orientation of the groups, then we would say that the ortho will slightly win. Here you guys can see the nitration of phenyl. Notice that here I have my nitric acid. I have my sulfuric acid. We know that we're going to generate what is it? It's going to be the Nitronium cation or nitronium ion. We know that OH is an ortho para director because it has a lone pair. That means that it's an electron donating. I mean it has two lone pairs. I'm just saying it has at least one lone pair. We know that it's going to direct either next to it, which is ortho, or at the bottom, which is para. But it turns out that in this case, ortho is slightly better. Why? Because if you make it ortho, then you can get a hydrogen bond between the phenyl and the nitrile group. If you make it para, it's less hindered. It's easier to add in that way but it can't interact with itself. It's not going to be quite as favored. This ratio actually isn't as big of a difference as you might think. It's actually only going to be a 60% to 35 percent ratio. Meaning that it's not even winning by that much but still this would be the only exception where we would expect more ortho. But for everything else, if asked to supply one major product, I mean you can always say it's a mixture. But if you're asked to supply one, you're usually going to go with the para product because it's the one that's less sterically hindered. All right, guys, so believe me, you don't need to know these percentages. I'm just giving them as a teaching moment so you guys can see actual examples in real life and you guys can understand how it plays out. But in general, you can just go with the rules that I'm telling you. All right, guys. Let's move on to the next video.
EAS:Ortho vs. Para Positions - Online Tutor, Practice Problems & Exam Prep
 Created using AI
Created using AIIn electrophilic aromatic substitution (EAS) reactions, ortho and para positions are favored by electron-donating groups. While ortho positions have two reactive sites, para positions are less hindered, often making them the major product. An exception occurs when hydrogen bonding favors the ortho product, as seen in nitration reactions. Generally, for a single major product, the para product prevails due to steric hindrance, except when self-hydrogen bonding occurs, leading to a slight preference for ortho products.
In general, we refer to the products of an EAS o,p-director as a mixture – but there are some patterns we can learn.

Ortho, Para major products
Video transcript
There is one exception to this rule – which occurs if the final product can hydrogen-bond with itself. Then, we would expect more ortho, than para product.
Do you want more practice?
More setsHere’s what students ask on this topic:
What is the difference between ortho and para positions in EAS reactions?
In electrophilic aromatic substitution (EAS) reactions, ortho and para positions refer to the locations on the benzene ring where substituents can attach. The ortho positions are adjacent to the substituent (positions 2 and 6), while the para position is directly opposite the substituent (position 4). Ortho positions have two reactive sites, making them statistically more likely to react. However, the para position is less sterically hindered, often making it the major product. The choice between ortho and para products depends on factors like steric hindrance and potential hydrogen bonding.
Created using AIWhy is the para product usually favored over the ortho product in EAS reactions?
The para product is usually favored over the ortho product in EAS reactions due to steric hindrance. The para position is further away from the electron-donating group, making it less crowded and easier for the electrophile to attack. Although there are two ortho positions compared to one para position, the reduced steric hindrance at the para position generally makes it the major product. An exception occurs when hydrogen bonding can stabilize the ortho product, as seen in some nitration reactions.
Created using AIWhat is the role of steric hindrance in determining the major product in EAS reactions?
Steric hindrance plays a crucial role in determining the major product in EAS reactions. Steric hindrance refers to the physical crowding around a reactive site, which can impede the approach of an electrophile. In the context of ortho and para positions, the para position is less sterically hindered because it is further away from the electron-donating group. This makes it easier for the electrophile to attack the para position, often resulting in the para product being the major product. However, if hydrogen bonding can occur, the ortho product may be favored.
Created using AICan hydrogen bonding influence the preference for ortho products in EAS reactions?
Yes, hydrogen bonding can influence the preference for ortho products in EAS reactions. When the final product can form hydrogen bonds with itself, the ortho product may be favored. This is because hydrogen bonding can provide additional stability to the ortho product. For example, in the nitration of phenol, the ortho product is slightly favored over the para product due to the ability to form a hydrogen bond between the hydroxyl group and the nitro group. However, this is an exception, and generally, the para product is favored due to reduced steric hindrance.
Created using AIWhat are blocking groups and how do they affect ortho and para positions in EAS reactions?
Blocking groups are substituents that are intentionally added to a benzene ring to control the position of subsequent substitutions in EAS reactions. They can either block or direct electrophiles to specific positions on the ring. For example, a bulky blocking group can be placed at the ortho position to prevent further substitution there, thereby directing the electrophile to the para position. Blocking groups are useful in synthetic chemistry for achieving selective substitution patterns on aromatic rings.
Created using AI
