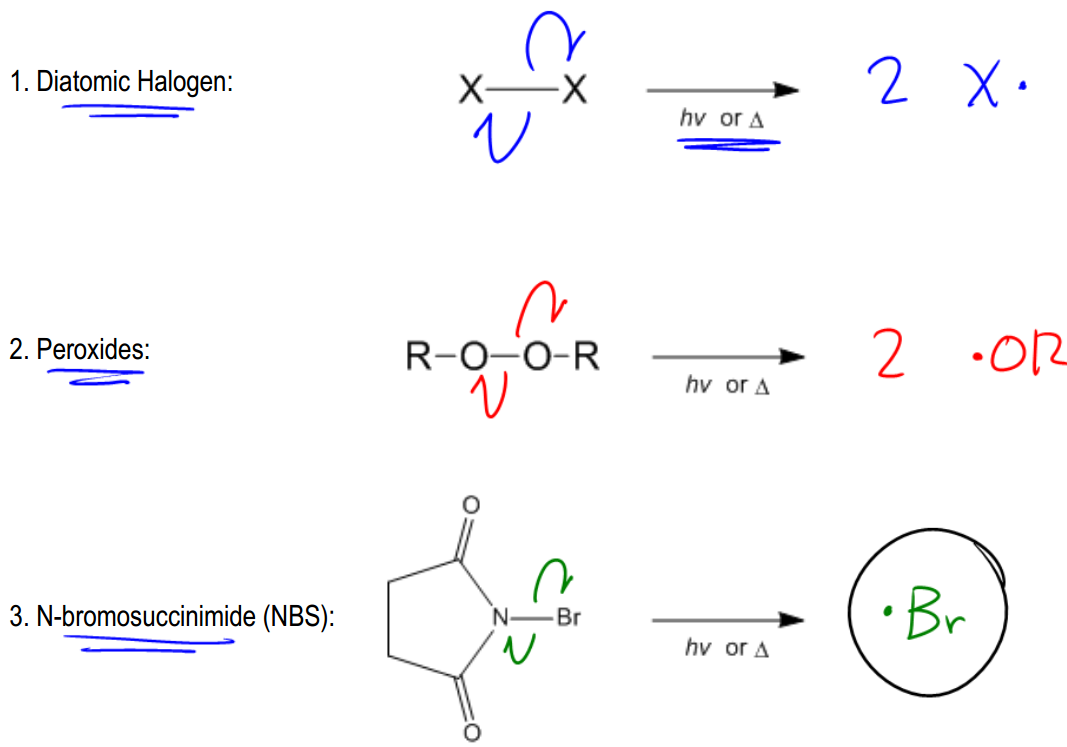
Radicals are highly energetic intermediates that exist for only brief moments in nature and within the human body. Understanding how these radicals are generated is crucial, as every radical reaction begins with a radical initiator. The formation of radicals occurs through a process known as bond cleavage, which can happen in two distinct ways: heterolytic and homolytic cleavage.
Heterolytic cleavage involves the breaking of a bond where both electrons from the bond are transferred to one atom, resulting in the formation of charged species—specifically, a cation and an anion. For example, when a carbon-halogens bond breaks heterolytically, the more electronegative atom (the halogen) attracts both electrons, leading to the creation of a positively charged carbon ion (C+) and a negatively charged halogen ion (X-). This process is characterized by the use of full-headed arrows to indicate the movement of both electrons.
In contrast, homolytic cleavage occurs when a bond breaks and each atom retains one electron, resulting in the formation of two radicals. This type of cleavage is indicated by half-headed arrows, also known as fish hook arrows, which signify the movement of a single electron. Homolytic cleavage typically requires higher dissociation energy compared to heterolytic cleavage, making it less common in organic reactions. However, it is essential for radical reactions, which are initiated by radical initiators.
In summary, understanding the differences between heterolytic and homolytic cleavage is fundamental in organic chemistry, particularly when studying radical reactions. Heterolytic cleavage leads to the formation of ions, while homolytic cleavage produces radicals, both of which play significant roles in various chemical processes.