Starting from the very beginning, radicals are these very high energy intermediates that in the natural world exist for only very short periods of time. If you ever hear about free radicals in nature or in your body, these are things that last for tiny amounts of time and they're gone. They terminate. So when we're dealing with reactions with radicals, the first thing we need to ask is how do we even get these radicals in the first place. So that brings us to radical initiators. Okay? So every radical reaction that we talk about is always going to start off with a radical initiator. So first of all, let's just talk about how radicals break off differently than regular single bonds. And basically, it turns out that single bonds can be broken in 2 different ways. They can be broken heterolytically or homolytically. So let me show you the difference between that. A normal heterolytic cleavage, that means that I'm breaking this bond and I'm going to get different charges on both sides, would mean that 2 electrons, both of the electrons from that bond are moving to 1 atom. That means that one atom is going to have a negative charge and one's going to have a positive. So let's look at this example bond right here. I have a carbon and some kind of halogen. How could we predict which of the species would get the negative charge or would get the lone pair on it? Do you guys know how to predict that? The way that we would predict is that we would say the one that's the most electronegative is the one that gets the electrons when the bond breaks. So there's actually a pretty powerful dipole going towards the X depending on which halogen we're using. So what we would say is that if we were to break this bond, the way we would break it is towards the X. Okay? So notice that I'm using a full arrow and that's showing that both electrons kind of pick up, pack their bags, and move to the X. Okay? What I wind up getting is ions. So I wind up getting a C+ and an X-. Okay? This is like I said, this would be heterolytic cleavage. Okay? Now, the reason that we call it heterolytic is because hetero stands for the word different. Okay? So you're getting different amounts of electrons on both. Okay? Now notice what this creates is ions. Okay? Your products are different ions, a cation and an anion. Okay? So that's one way to break bonds. Okay? But another way to break them is that I could break them just taking one electron from each side. So I could take one electron and give it to that X. I could take another electron and give it to that X. Now notice that one thing that was different about this bond than the other one was that there was really no dipole. I couldn't tell which one was more electronegative or not because they both had the same electronegativity. So that's actually going to be important. Okay? What that's going to do is it's going to give me instead of a negative and a positive, it's gonna give me 2 of the same thing. Hence, the name homolytic cleavage. Okay? In this case, homo meaning same, that you're getting basically the same electrons on both. And notice that our product here would be radicals. All right. Cool so far? Awesome. So, basically, I want to show you guys the difference between the arrows that I just drew. When we want to draw that 2 electrons are moving to an atom, we say that full curved arrows are used to indicate the movement of 2 electrons. That means it's a full headed arrow. It has both sides of that arrow head. Okay? When we want to only show that one electron is moving, we would use a half headed arrow or what is sometimes called a fish hook arrow because it's only got half of the arrowhead on it. Okay? Like I used on the X's. Okay? So it turns out that homolytic dissociation is usually much higher dissociation energy. It's typically higher than corresponding heterolytic dissociation energy. So what that means is that most of the time when we're breaking bonds in organic chemistry, we're actually going to be using the blue method, the one that's heterolytic. Okay? And you're going to see that whether you get into other types of reactions or just later on in the course. You're going to see that we're going to use a lot of heterolytic cleavage. Okay? Homolytic cleavage is really reserved just for radical reactions. Okay? And these are reactions that are favorable for just a small set of reasons, and it always starts off with an initiator.
Radical Reaction - Online Tutor, Practice Problems & Exam Prep
 Created using AI
Created using AIRadicals are high-energy intermediates that exist briefly in reactions, initiated by radical initiators. Bonds can break heterolytically, forming ions, or homolytically, producing radicals. Homolytic cleavage occurs in weak bonds, such as diatomic halogens, peroxides, and N-Bromosuccinimide (NBS), often requiring heat or light. This process is crucial for radical reactions, where one electron from each atom is transferred, creating radicals that can further react. Understanding these mechanisms is essential for grasping organic chemistry reactions and their applications.
Radical reactions require an initial first step to get going. We call this reagent the radical initiator.
Heterolytic vs. Homolytic Bond Cleavage .
Video transcript
Chemical bonds can be cleaved in two ways:Heterolytically (ionic cleavage) and homolytically (radical cleavage).

Homolytic dissociation energy is much higher than a corresponding heterolytic dissociation energy.
What are Radical Initiators?
Video transcript
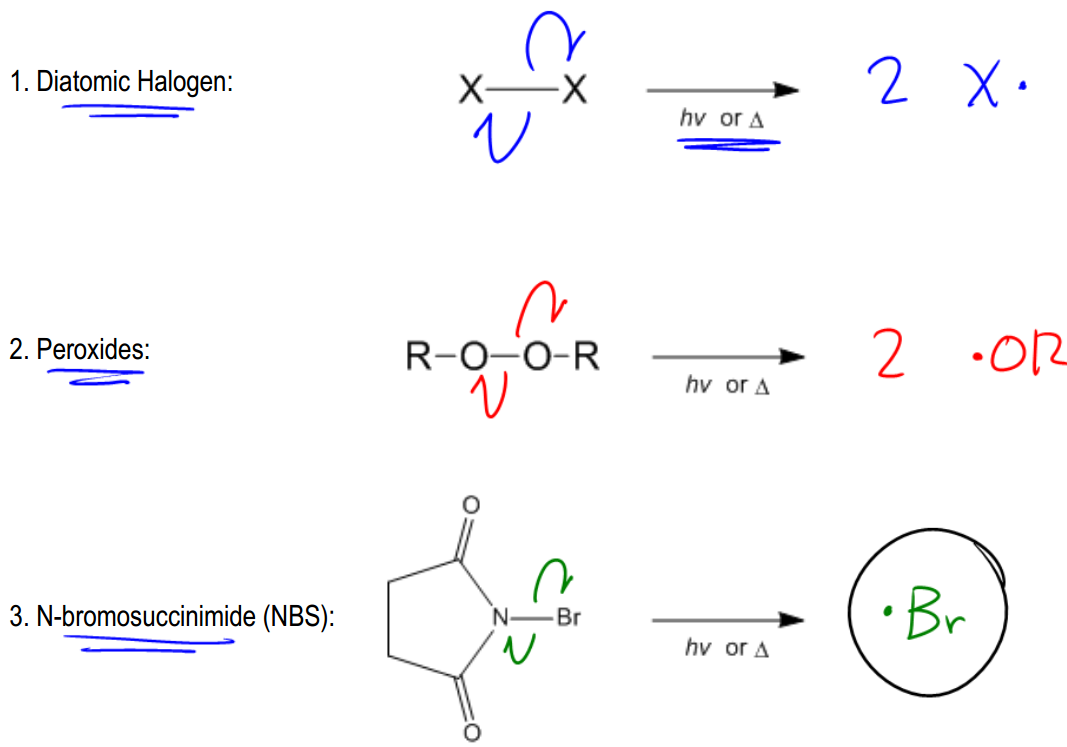
So let's go ahead and see how this works. Basically, it turns out that not that many like I said, it takes a lot of energy to break the bond, so that only one electron goes to each. What that means is that in order to get a radical initiator, we should actually start off with molecules that have relatively weak bonds. Okay? If they have relatively weak bonds, it will be easier to split them off homolytically. Okay? If it's a very, very strong bond, forget it. That's going to be difficult to start off with making a radical. Okay? So it turns out that there are 3 really common reagents that have weak bonds that are able to easily make these radicals. Okay? And what that is is diatomic halogen, k, peroxides, and something called NBS. Okay. I'm going to start off with the easiest one then we'll work our way down. Okay? So diatomic halogen, this is actually depending on which halogen you use, this is actually one of the weaker bonds in organic chemistry. Okay? And what that means is that in the presence of heat or light, okay, you need heat or light. Why? Because like I said, it takes a lot of energy to break these bonds so that one electron goes to each atom, you need some kind of energy source to make this happen. Okay? We usually use ultraviolet light or heat. What that's going to do is it's going to excite the electrons in that bond and enable them to jump homolytically onto each atom. Okay? So in the presence of heat or light, what we're going to wind up getting is 2X• radicals. Okay? That's called a radical initiator because now I have my first radical that I can use for the mechanism. We don't know the mechanism yet, but at least you know this is how you can get a radical. Cool, right? By the way, the arrows would just look like the ones that I drew. One electron from the 2 electrons in the bond, one electron jumps to each x. Okay? The same thing can be said of peroxides. Peroxides also have a relatively weak bond. That OO bond is relatively weak. And peroxides are going to be able to, once again, break off homolytically, and in this case, it would make 2OR• radical initiators. Okay? So once again, I'm using a relatively weak bond in the presence of heat or light and that's going to dissociate homolytically and what I wind up getting is radicals. Okay? Lastly, we have something called NBS. Now, you guys aren't really going to understand why this is called N Bromosuccinimide until Orgo 2. In orgo 2, we actually will understand why how to name that. But for right now, all I want you guys to be able to do is recognize the full name, recognize the short name, which is the acronym, NBS, which is the way that most professors refer to it. But you should also recognize the structure just in case your professor wants to pull a fast one on you and test your knowledge. Okay? And it turns out that NBS has again a pretty weak bond between that nitrogen and that bromine, So it turns out that NBS is actually a source of bromine radicals. And in the presence of heat or light, you'd get one electron moving to each. The part that we care about is that you're going to get 1Br• radical. Okay? In fact, we're not even going to draw the other side because the other side is not really involved in reactions. All I care about is that I'm getting this one Br• radical that can then react with other things. Alright? So I hope that you guys are understanding from the very beginning, we always need an initiator. We need something that's going to make this improbable type of cleavage happen and that's through these weak bonded elements like this. Okay? And now that we understand this, we can move on through the mechanism. So let's keep going. Let me know if you have any questions.
Radical initiators have relatively weak bonds that can be more easily cleaved by hemolysis.
Do you want more practice?
More setsHere’s what students ask on this topic:
What are radical initiators and why are they important in radical reactions?
Radical initiators are compounds that can easily form radicals through homolytic cleavage, which is the breaking of a bond such that each atom retains one electron. They are crucial in radical reactions because they generate the initial radical needed to propagate the reaction. Common radical initiators include diatomic halogens, peroxides, and N-Bromosuccinimide (NBS). These compounds have relatively weak bonds that can be broken with heat or light, providing the energy required to form radicals. Without radical initiators, the high energy required to form radicals would make these reactions impractical.
Created using AIWhat is the difference between heterolytic and homolytic bond cleavage?
Heterolytic bond cleavage occurs when a bond breaks and both electrons from the bond move to one of the atoms, resulting in the formation of a cation and an anion. This type of cleavage is common in many organic reactions. Homolytic bond cleavage, on the other hand, involves the equal splitting of the bond, with each atom retaining one electron, forming two radicals. Homolytic cleavage requires more energy and is typically seen in radical reactions. The type of cleavage is indicated by the arrows used: a full-headed arrow for heterolytic and a half-headed (fish hook) arrow for homolytic cleavage.
Created using AIWhy do radical reactions require heat or light to proceed?
Radical reactions require heat or light because these energy sources provide the necessary activation energy to break the weak bonds in radical initiators homolytically. For example, diatomic halogens, peroxides, and N-Bromosuccinimide (NBS) have relatively weak bonds that can be broken when exposed to ultraviolet light or heat. This energy excites the electrons, allowing the bond to split evenly and form radicals. Without this energy input, the homolytic cleavage needed to generate radicals would not occur, and the radical reaction would not proceed.
Created using AIWhat are some common radical initiators used in organic chemistry?
Common radical initiators used in organic chemistry include diatomic halogens (such as Cl2 and Br2), peroxides (such as hydrogen peroxide, H2O2, and benzoyl peroxide), and N-Bromosuccinimide (NBS). These compounds have relatively weak bonds that can be broken homolytically in the presence of heat or light, generating radicals. For instance, diatomic halogens can form two halogen radicals, peroxides can form two alkoxy radicals, and NBS can form bromine radicals. These radicals then initiate further reactions by reacting with other molecules.
Created using AIHow does the bond strength of a molecule affect its ability to act as a radical initiator?
The bond strength of a molecule significantly affects its ability to act as a radical initiator. Molecules with weaker bonds are more likely to undergo homolytic cleavage, which is necessary to form radicals. For example, diatomic halogens, peroxides, and N-Bromosuccinimide (NBS) have relatively weak bonds that can be broken with the input of heat or light. Stronger bonds require more energy to break, making them less suitable as radical initiators. Therefore, the weaker the bond, the more easily the molecule can generate radicals and initiate radical reactions.
Created using AIYour Organic Chemistry tutors


- Using the given starting material and any necessary organic or inorganic reagents, indicate how the desired co...
- Using the given starting material and any necessary organic or inorganic reagents, indicate how the desired co...
- Starting with cyclohexane, how could the following compounds be prepared? c.
- Starting with cyclohexane, how could the following compounds be prepared? b.
- Design a multistep synthesis to show how the following compounds can be prepared from the given starting mater...
- Design a multistep synthesis to show how the following compounds can be prepared from the given starting mater...
- Show how the following compounds could be prepared from 2-methylpropane: c. 2-iodo-2-methylpropane
- Using cyclohexane as one of your starting materials, show how you would synthesize the following compounds.(e)...
- Show how you would accomplish the following synthetic conversions.d. 2−methylbutan-2-ol -> 2-bromo-3-methyl...
- Identify A through O:
- Identify A–J:
- Identify A–J:
- Show how the following compounds could be prepared from 2-methylpropane:b. 2-methyl-1-propene
- The cationic polymerization of isobutylene (2-methylpropene) is shown in [SECTION 8-16A] <IMAGE>. Isobut...
- Show how you would convert (in one or two steps) 1-phenylpropane <IMAGE> to the three products shown bel...