Hey guys. In this video, I want to discuss enolate alkylations and acylations. There's really nothing tricky to this reaction. As long as you can draw an enolate, you can do an enolate alkylation because all it is is that you're either exposing an enolate to an alkyl halide or you're exposing an enolate to an acid chloride. What happens is the base grabs one of the protons on the alpha carbon. You make a negatively charged enolate and that does a nucleophilic attack on either the alkyl halide or on the acid chloride. If you use an alkyl halide, then this would be my product, an alpha-alkylated carbon. If you use your acid chloride, then you're going to wind up getting an acylated carbon. You'd get a beta-dicarbonyl with an R group depending. That's it. This is a very easy mechanism to understand. Very easy reaction to understand. Let's move on to the next part of the page.
Enolate Alkylation and Acylation - Online Tutor, Practice Problems & Exam Prep
 Created using AI
Created using AIEnolate alkylations and acylations involve exposing an enolate ion to an alkyl halide or acid chloride, resulting in alpha alkylated or acylated products. For asymmetrical ketones, directed reactions utilize different bases to control enolate formation, distinguishing between thermodynamic and kinetic products. The thermodynamic product is the more stable enolate, while the kinetic product is formed more easily. When working with esters, LDA is the preferred non-nucleophilic base to avoid unwanted reactions like hydrolysis or transesterification, ensuring successful alkylation or acylation.
General Reaction
Video transcript
Directed Reactions
Video transcript
This brings us to directed reactions because the mechanism that I showed you above, even though it's great and very easy to use, it only works with symmetrical ketones. It only works when there's only one type of enolate possible. But what happens if you have an asymmetrical ketone? That means that you would theoretically have more than one enolate possible. For example, take this ketone into consideration. I could get the red enolate on the more substituted side or I could get the blue enolate on the less substituted side. Would I get both? Would I get R groups forming on both sides? No. This is something that we have to answer using directed reactions. It turns out that you can use different bases to direct the direction of the deprotonation to make the enolate. This is a concept that should be familiar to you guys because we've used it before. This is simply a thermodynamic versus kinetic control reaction. The thermodynamic product, let's just review, is going to be the one that's more stable. In this case, since this is an enolate intermediated reaction, it's going to be the one that has the lowest overall energy or the most stable enolate. I'll show you how to determine which one's more stable in a second.
The kinetic product is the one with the lowest activation energy. How do we know which enolate is stable? It's more stable. Remember that an enolate goes through 2 resonance structures. One of the resonance structures is like this. But another resonance structure looks like this. Well, look at that. One of the resonance structures has an alkene in it. The way you determine which one is the most stable enolate is by the most substituted alpha carbon. Why? I'm drawing on that side and I really should have drawn it on the side with the actual red one. That's going to be over here. If you want, I know you hate me right now, but you can pause the video and redraw that on this side if you want or you can just draw an arrow. The one with the most substituted alpha carbon is going to be the most stable. That means that the red one would be my thermodynamic control because the one that is going to it has the most R groups on the alpha carbon. That means it's going to have the most R groups on my double bond and that's what makes the double bond stable. Remember that double bonds are stabilized through R groups.
What makes a kinetic enolate is the easiest to make or that's going to be the least substituted because it's less sterically hindered. These are the competing themes here. Specifically, how do we choose one or the other? If you want the blue enolate, you choose a bulky base like LDA. LDA is the most popular one in this section. You could also use tert-butoxide. A bulky base is going to favor the kinetic product because it's the easiest one to make. Whereas a small base, so for example, NaOH, is going to favor the thermodynamic because it's not going to have trouble getting into that spot to deprotonate and it's going to make the most stable enolate overall. You would determine which side you substitute by looking at your base and then using that enolate to react with your electrophile, whether it be an alkyl halide or an acid chloride. Awesome. In the next video, I want to talk about how this applies to esters.
Enolates of Esters
Video transcript
Really quickly, esters can also be alkylated and acylated. The only catch is that you need to use LDA. You can't use any other base. You can't just use hydroxide. Why do you think it would be a mistake to use OH- on an ester? Can you think of a reason? If you use OH- on an ester, what you're actually going to get is a hydrolysis. You're going to wind up getting a carboxylate. That would be like an SN2 reaction. You don't want your base to react with the alkyl group, same as if I used let's say OR1. If I used an alkoxide base with an R different from the R in my alkyl group, I'm going to get a transesterification. I don't want that to happen. What we do is we use a non-nucleophilic. LDA is considered a non. Let me draw it for you really quick.
LDA, in case you haven't seen it in a while: nitrogen, isopropyl, isopropyl, lithium, this is considered a non-nucleophilic base. Why? Because it has a very hard time can't donate its electrons. Can you see that? Perfect. Sorry, my handwriting sucks down there. But LDA is a non-nucleophilic base. It can't donate its electrons, so it's not going to be able to attack this carbon. Instead, it just reacts as a base and pulls off an alpha hydrogen. By using LDA, we strategically make the enolate and we don't have to worry about any types of carboxylic acid derivative reactions.
By the way, if you haven't studied carboxylic acids yet or if that wasn't your forte, don't worry about that. I don't really care that you know all the details here. If this is the first time you're hearing about transesterification or SN2 or any of that, just focus on the point. The point being, use LDA. For reasons that you don't understand yet, use LDA, and later on, when you get to carboxylic acid derivatives, it will make more sense. LDA makes an enolate, and then you would then do your attack on the alkyl halide or the acid chloride and you would get your products either OR with an R group OR OR with an acyl group. Make sense? Awesome. Pretty straightforward page. Nothing too crazy here. Let's move on to the next page.
Determine if the reaction is thermodynamically or kinetically controlled
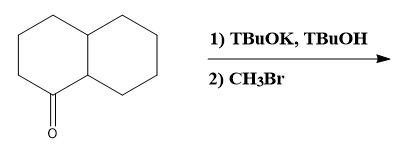
Do you want more practice?
More setsHere’s what students ask on this topic:
What is enolate alkylation and how does it work?
Enolate alkylation involves the reaction of an enolate ion with an alkyl halide to form an alpha-alkylated product. The process begins with a base deprotonating the alpha carbon of a carbonyl compound, creating a negatively charged enolate ion. This enolate then acts as a nucleophile, attacking the electrophilic carbon in the alkyl halide, resulting in the formation of a new carbon-carbon bond at the alpha position. The general mechanism can be summarized as:
Created using AIWhat is the difference between thermodynamic and kinetic enolate formation?
Thermodynamic and kinetic enolate formations differ in stability and ease of formation. The thermodynamic enolate is the more stable product, typically formed at higher temperatures and with small bases like NaOH. It is characterized by having the most substituted alpha carbon, which stabilizes the enolate through hyperconjugation and inductive effects. The kinetic enolate, on the other hand, is formed more quickly and at lower temperatures using bulky bases like LDA (lithium diisopropylamide). It is less substituted and forms faster due to lower steric hindrance. The choice between these two depends on the desired product and reaction conditions.
Created using AIWhy is LDA used for enolate formation in esters?
LDA (lithium diisopropylamide) is used for enolate formation in esters because it is a strong, non-nucleophilic base. Using LDA prevents unwanted side reactions such as hydrolysis or transesterification, which can occur with other bases like hydroxide or alkoxides. LDA deprotonates the alpha carbon of the ester, forming the enolate without reacting with the ester itself. This ensures that the enolate can then react with an alkyl halide or acid chloride to form the desired alkylated or acylated product without complications.
Created using AIHow do you control the formation of enolates in asymmetrical ketones?
In asymmetrical ketones, the formation of enolates can be controlled by choosing the appropriate base. For thermodynamic control, which favors the more stable enolate, a small base like NaOH is used. This base can easily access the more substituted alpha carbon, leading to the formation of the more stable enolate. For kinetic control, which favors the less substituted and more easily formed enolate, a bulky base like LDA or tert-butoxide is used. These bulky bases preferentially deprotonate the less sterically hindered alpha carbon, resulting in the kinetic enolate. The choice of base thus directs the formation of the desired enolate.
Created using AIWhat are the products of enolate acylation?
Enolate acylation involves the reaction of an enolate ion with an acid chloride, resulting in the formation of a beta-dicarbonyl compound. The process begins with a base deprotonating the alpha carbon of a carbonyl compound, creating a negatively charged enolate ion. This enolate then acts as a nucleophile, attacking the electrophilic carbon in the acid chloride. The resulting product is a beta-dicarbonyl compound, which has two carbonyl groups separated by a single carbon atom. The general mechanism can be summarized as:
Created using AIYour Organic Chemistry tutors


- The following are all substitution reactions, two of which we study in later chapters. With no knowledge of me...
- Give common names for the following compounds. (a) (b)
- A carboxylic acid is formed when an a-haloketone reacts with hydroxide ion. This reaction is called a Favorski...
- How could each of the following compounds be prepared from a ketone and an alkyl halide? b.
- How could each of the following compounds be prepared from a ketone and an alkyl halide? a.
- Show how the following ketones might be synthesized from the indicated acids, using any necessary reagents. (...
- Predict the products of the following reactions. (c)
- Predict the major products of the following reactions. (a) < of reaction> (b) < of reaction> (c) &...
- How could each of the following compounds be prepared from cyclohexanone?b. <IMAGE>
- How could each of the following compounds be prepared from cyclohexanone?a. <IMAGE>
- Draw the products of the following reactions: c. acetone + LDA/THF: (1) slow addition of ethyl acetate; (2) HC...
- How could you prepare the following compound using a starting material that contains no more than three carbon...
- Draw the products of the following reactions: c.
- Show how 4-methyl-3-hexanol can be synthesized from 3-pentanone.
- Show how you would accomplish the following multistep conversions. You may use any additional reagents you ne...