So it turns out that alkenes are stabilized by a force called hyperconjugation. Okay? So let's go ahead and write this down, hyperconjugation. And as you guys have already learned or will learn, hyperconjugation is a force that also stabilizes carbocations. So in fact, when we talk about alkenes, many times we'll talk about carbocations hand in hand because it's the same exact force that's stabilizing both of them. So how does this work? Basically, what it means is that in a double bond, I have a pi bond that's being formed by 2 overlapping p orbitals. Okay? That pi bond can be stabilized by having sigma bonds that are close by share electrons with it. Okay? And that's exactly what happens in hyperconjugation. So as you'll notice, what I have here is a double bond overlapping on the top and the bottom, and then what I have is an adjacent sigma bond right here, between the carbon and the H. And it turns out that the more groups that I have, the more hydrogens I have overlapping their bonds with the bonds from the pi bond, the more stable that pi bond is going to be. Because these electrons are going to be able to share and basically donate a little bit of their density to the double bond. Alright? So what that means is that the more r groups that I have around my double bond, the more stable it's going to be. Alright? And that's exactly going to be the trend that we use to determine the most stable alkene. So basically, since this phenomenon of hyperconjugation is only possible with r groups, the more substituted the alkene, the more stable it is. Okay? And that leads us to the following trend. The following trend of alkene stability just has to do with how many R groups can they pile around a double bond. So as you can see, the best kind of double bond possible would be called tetra substituted. Okay? Why? Because it's just 4 groups. That means I have the maximum number of r groups right around my double bond. As I start taking R groups away and replacing them with H, that's gonna reduce the amount of hyperconjugation that can stabilize my double bond. So as you can see trisubstituted would be next, then di, then mono, and then finally the worst, so sad face over here, is unsubstituted. Because unsubstituted can't hyperconjugate at all. There's nothing that can donate electrons to that pi bond, so it's pretty much just going to be unstable. Okay? Now it turns out that for the purposes of disubstituted, there's actually a few different ways that we can order those R groups. So let's go ahead and look into that more. Basically, when you have a disubstituted double bond, you have options. It's not like you're just going to have one type of disubstituted. You could have them where both r groups are facing the same side of the double bond, that would be cis. You could also orient them so that both sides are facing opposite sides, that would be trans. And then finally, you could orient it so that both of the r groups are on the same corner of the double bond. They're actually coming off the same carbon. And this is a word called gem, which stands for the word geminal. Okay? Just so you guys know, geminal is a position word. It's actually a word that we'll use more in Orgo 1 and Orgo 2 later. But all it means is that I have two things coming off the same carbon. The way that I like to think about it is geminal is like the word Gemini and Gemini means twins. Right? So it's like you have 2 things coming off the same carbon. These r groups are like twins. They're both coming off the same carbon. Okay? So for whatever reason it is, geminal is going to be most stable, then trans is going to be more stable than then well, trans is going to be the second most stable and then cis is going to be the least stable. Now the pattern between cis and trans is really easy to understand because cis, these groups are kind of interfering with each other. They're in each other's space. Whereas trans, they're facing opposite to each other, so they're more stable, there's more room to breathe. Now, why geminal is more stable than trans? I'm not exactly sure, but it's just something that you guys can memorize and you guys can know it for your test. Alright? So here's a really easy question just following up on what we just talked about. We have 4 alkenes here. Go ahead and try to rank them in order and of stability, and then when you're done I'll go ahead and answer it.
Alkene Stability - Online Tutor, Practice Problems & Exam Prep
 Created using AI
Created using AIAlkenes are stabilized by hyperconjugation, a force that also stabilizes carbocations. The stability of alkenes increases with the number of alkyl (R) groups around the double bond, following the trend: tetra-substituted > tri-substituted > di-substituted > mono-substituted > unsubstituted. In disubstituted alkenes, configurations can be cis, trans, or geminal, with geminal being the most stable, followed by trans and then cis due to steric hindrance. Understanding these stability trends is crucial for predicting alkene reactivity and behavior in organic synthesis.
Not all alkenes were created equal. Like carbocations, alkenes are stabilized through a phenomenon called hyperconjugation.
Hyperconjugation allows adjacent -R groups (mostly C-C and C-H σ-bonds) to create shared molecular orbitals with π-bonds, stabilizing the bond.
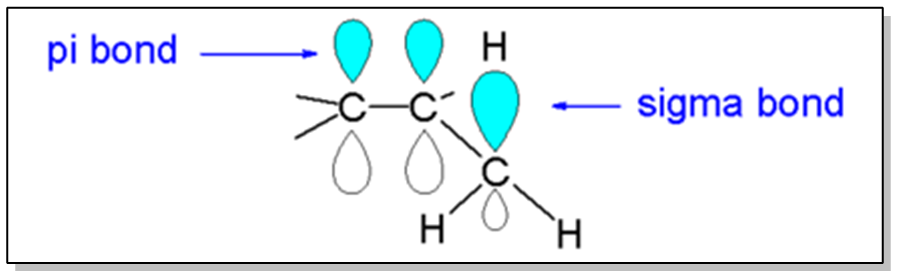
Understanding trends of alkene stability.
Video transcript
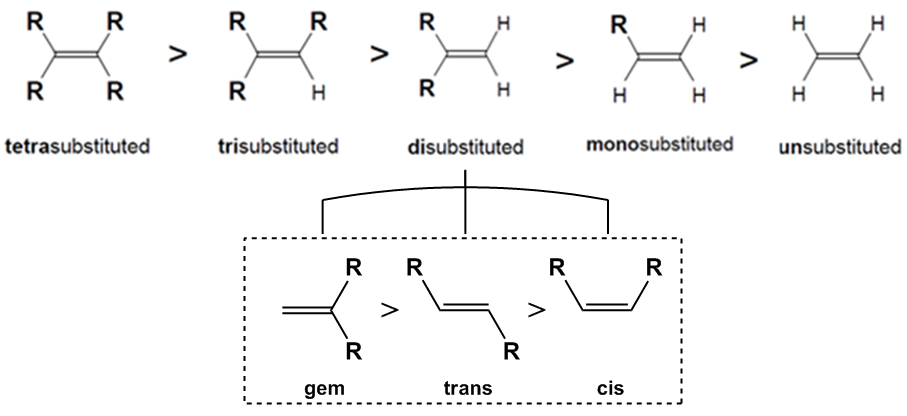
So basically, the more –R groups attached to the double bond, the more stable the double bond will be.
Specifically, when it comes to di-substituted bonds, the order os stability is gem > trans > cis.
- trans > cis due to steric hindrance. Groups have less freedom of movement in the cis position.
- gem > trans due to better hyperconjugation. The full explanation for this trend is beyond the level of this course. Just memorize it.
Rank the following alkenes in order of lowest to highest heat of combustion.
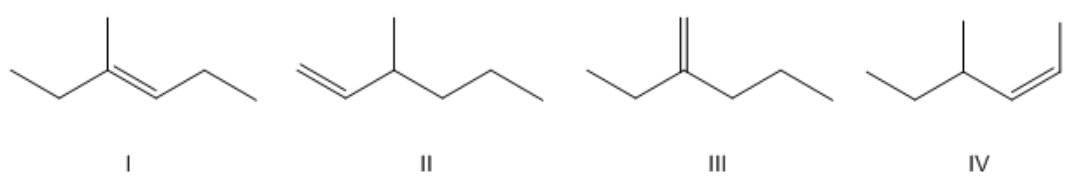
Heat of Combustion
Video transcript
Alright, so the first place we needed to start was figuring out what is lowest to highest heat of combustion. So remember earlier when we talked about heat of combustion, what heat of combustion has to do with is it's a measure of energy. So if you have a high heat of combustion, a high enthalpy, then what that means is that you have high energy. If you have high energy, that means you have low stability. So if it's saying here in order of lowest to highest heat of combustion, that means I want to start with the lowest, which is going to be the most stable, and end with the highest, which is going to be the least stable. Does that make sense? So even before this question began, there was a little bit of thinking that you needed to do. Now that we have that figured out, let's go ahead and order these guys. So we know that there's definitely going to be a winner here and that winner is going to be number 1. Why is that? Because if we identify the types of substituted double bonds we have, this one is trisubstituted because my double bond doesn't count and it has 3 branches coming off of it. 1, 2, 3. So, 3 yellow things. Let's just call it like that, really easy. Okay? 2 is going to be monosubstituted. Why? Because if I circle a double bond, I'm not including that, I only have one branch coming off of it, so one yellow area. So that would be mono. We know that's not going to be very good. This one would be disubstituted because I have 2 branches, 1, 2 di. And then this one would also be... oops, I did that wrong. This one would also be disubstituted because I have 2 branches, one here and one here. So now all I have to do is I have to figure out which of these is the most stable and which of these is the least. I know the most stable is going to be number 1 because that's trisubstituted. And I also know that the least stable is going to be number 2 because that one is monosubstituted. The hard part comes between numbers 3 and 4 because both of these are disubstituted. So how do I tell which one is better? The one that's better is going to be the one that is geminal and the one that's worse is going to be the one that is cis. Because for the cis one, both of my groups are facing the same side of the double bond, so that's really bad. For the geminal, they're facing on the same carbon, which is actually good. So what that means is that 3 is going to be more stable than 4, but still, 4 is going to be more stable than 2 because 4 is actually disubstituted and 2 is only monosubstituted. So in that case, cis is better than cis... disubstituted is better than just monosubstituted. Alright guys, so that's a really basic concept. This is an easy question on your exam. Most likely you will get a question like this. So this should be 3 points for you guys. Alright? Hope that made sense. Let's go ahead and move on to the next topic.
Do you want more practice?
More setsHere’s what students ask on this topic:
What is hyperconjugation and how does it stabilize alkenes?
Hyperconjugation is a stabilizing interaction that occurs when sigma bonds (usually C-H or C-C) adjacent to a double bond or carbocation overlap with the empty p-orbital or π-orbital. In alkenes, the π-bond formed by overlapping p-orbitals can be stabilized by adjacent sigma bonds sharing their electron density. This sharing of electron density helps to delocalize the electrons, thereby stabilizing the double bond. The more alkyl (R) groups around the double bond, the more hyperconjugation can occur, leading to increased stability of the alkene.
Created using AIHow does the number of alkyl groups affect alkene stability?
The stability of alkenes increases with the number of alkyl (R) groups attached to the double bond. This is due to hyperconjugation, where adjacent sigma bonds share electron density with the π-bond, stabilizing it. The trend in stability is as follows: tetra-substituted (most stable) > tri-substituted > di-substituted > mono-substituted > unsubstituted (least stable). More alkyl groups mean more hyperconjugation, leading to greater stability.
Created using AIWhat is the difference between cis, trans, and geminal disubstituted alkenes in terms of stability?
In disubstituted alkenes, the stability varies based on the arrangement of the substituents. Geminal alkenes, where both substituents are on the same carbon, are the most stable. Trans alkenes, with substituents on opposite sides of the double bond, are the next most stable due to reduced steric hindrance. Cis alkenes, with substituents on the same side, are the least stable because of increased steric hindrance between the groups.
Created using AIWhy are tetra-substituted alkenes more stable than mono-substituted alkenes?
Tetra-substituted alkenes are more stable than mono-substituted alkenes because they have more alkyl (R) groups around the double bond. These additional R groups allow for more hyperconjugation, where adjacent sigma bonds share electron density with the π-bond, stabilizing it. The increased electron delocalization from more R groups leads to greater stability in tetra-substituted alkenes compared to mono-substituted ones.
Created using AIHow does steric hindrance affect the stability of cis and trans alkenes?
Steric hindrance significantly affects the stability of cis and trans alkenes. In cis alkenes, the substituents are on the same side of the double bond, leading to increased steric hindrance as the groups are closer together and interfere with each other. This makes cis alkenes less stable. In trans alkenes, the substituents are on opposite sides of the double bond, reducing steric hindrance and making them more stable compared to cis alkenes.
Created using AIYour Organic Chemistry tutors


- Rank the following compounds from most stable to least stable: trans-3-hexene, cis-3-hexene, cis-2,5-dimethyl...
- The energy difference between cis- and trans-but-2-ene is about 4 kJ/mol; however, the trans isomer of 4,4-dim...
- For each set of isomers, choose the isomer that you expect to be most stable and the isomer you expect to be l...
- Explain why each of the following alkenes is stable or unstable. a. 1,2-dimethylcyclopentene b. trans-1,2-dime...
- A double bond in a six-membered ring is usually more stable in an endocyclic position than in an exocyclic pos...
- Using [TABLE 7-2] as a guide, predict which member of each pair is more stable, as well as by about how many ...
- Which species in each pair is more stable? f.
- Which species in each pair is more stable? a.CH3C−HCH2CH3 or CH3CH2CH2C−H2
- Conjugated dienes, molecules containing two alkenes separated by one single bond, are discussed in detail in C...
- Conjugated dienes, molecules containing two alkenes separated by one single bond, are discussed in detail in C...
- (••••) LOOKING AHEAD Bromination of buta-1,3-diene with a single equivalent of Br₂ can give either of two pro...
- Rank each group of compounds in order of increasing heat of hydrogenation. (b)
- Rank each group of compounds in order of increasing heat of hydrogenation. (a) hexa-1,2-diene; hexa-1,3,5-tri...
- (•••) LOOKING AHEAD Consider the Cope rearrangement, a reaction we describe in Chapter 20. (a) Using the knowl...
- Rank the following alkenes in order of stability ( 1 = most stable; 5 = least stable ). Explain your order. ...
- The same alkane is obtained from the catalytic hydrogenation of both alkene A and alkene B. The heat ofhydroge...
- a. Which is the most stable: 3,4-dimethyl-2-hexene, 2,3-dimethyl-2-hexene, or 4,5-dimethyl-2-hexene?b. Which c...
- Of the compounds you named in Problem 45:a. Which is the most stable?a. <IMAGE>b. <IMAGE>c. <IM...
- Of the compounds you named in Problem 45:b. Which is the least stable?a. <IMAGE>b. <IMAGE>c. <I...
- Which species in each pair is more stable?c. C−H2CH2CH═CH2 or CH3C−HCH═CH2d. <IMAGE>
- Use the data in [TABLE 7-2] <IMAGE> to predict the energy difference between 2,3-dimethylbut-1-ene and 2...
- Explain why each of the following alkenes is stable or unstable.h. <IMAGE>i. <IMAGE>
- For each set of isomers, choose the isomer that you expect to be most stable and the isomer you expect to be l...
- For each set of isomers, choose the isomer that you expect to be most stable and the isomer you expect to be l...
- In 1935, J. Bredt, a German chemist, proposed that a bicycloalkene could not have a double bond at a bridgehea...