Hey everyone. So, in this video we're going to take a look at wave functions. But before we get to them, let's talk about the importance of quantum mechanics. So, why is quantum mechanics an important topic? Well, what we need to understand is that the smaller an object gets, the more likely it can behave as either a particle or a wave, and this comes with great complications in terms of determining the location of that particle, how it's moving, etc. Quantum mechanics states that electrons, because they are so small, behave both as particles and as waves. With quantum mechanics, we have the Heisenberg uncertainty principle which states that we cannot simultaneously know an electron’s speed and position. So, we might know the speed at which an electron is traveling, but we won’t know its position. Or we may know its position, but not know how fast it's moving. Because of this issue, we more focus on the probability of an electron’s location, and this is where wave functions come in handy. So, we're going to say equations called wave functions correspond to the energy state of an electron. And to symbolize this wave function, we use the symbol of Psi. Okay. Wave functions are corresponded by this image of a Psi. And we're going to say the relative probability of finding an electron can be derived from the wave function, and we do this by squaring our Psi. Okay. So here again we don't know both the speed and position of an electron, so we're talking about the probability of finding an electron's location instead. Now, we're going to say here that the three-dimensional plot of the y squared, so relative probability, is called an atomic orbital. Coming from general chemistry, we know that an orbital is the probable location of any defined electron. So, this is our chance of finding electrons where it's going to be high. Remember from general chemistry we talked about different types of orbitals. So, we have s orbitals, which are spherical in nature like 1s and 2s, and then we have p orbitals, which look like dumbbells. But an easier way to remember the shape of a p orbital, so p here, p knot, also starts with a p. These kind of look like peanuts. Okay. So that’s a good way to help you remember the shape of a p orbital. Now remember when we talk about these different orbitals, they're found within different energy levels. So, when we're looking at, let's say, the carbon atom, we could talk about how can we display its orbital diagram. Remember for carbon, its atomic number is 6, which means it has 6 electrons, so that means its electron configuration is 1s2, 2s2, 2p2. Now, let's start filling out each one of these orbitals. First of all, remember an s orbital has only 1 orbital, its shape is spherical in nature. Remember that each one of these orbitals, these boxes, can house a maximum of 2 electrons. Following what we call the Pauli exclusion principle, we're not going to go too much into those things. These are simple things that we learned in general chemistry. Remember the electrons in there have to have opposite spins. So we'd have 1 electron spinning up and then we have 1 electron spinning down. Now the order doesn't really matter which one you write first, they just have to have opposite spins. Once we filled up the 1s we move on to 2s. 2s has more energy and we move from lower energy orbitals to higher energy orbitals following Aufbau's principle. So, here we go 1 up, 1 down. Now the 2ps, there are 3 of them: x, y, and z. Okay. They all have the same energy since they're all 2p orbitals. The only difference is the position in which they lie on the x-axis, the y-axis, and the z-axis. But they're all the same energy. Because of this, you could technically start filling them up at pz if you want, but traditionally we fill them out starting with px. And we're going to say here, following our half-spin rule, so Hund's rule, we half-fill first, we have to fill in 2 electrons. So one up, one up. You could also do 1 down, 1 down. That would also work as well, but traditionally, start out with the one spinning up first. Now if we look at this, this is how we complete the orbital diagram for the carbon atom. So, this tells us that in the first shell, which only houses the 1 s orbital, we have a maximum of 2 electrons possible. And in the second shell, we have both 2s and 2p orbitals. This, in theory, can hold up to 8 electrons. It doesn't hold 8 for a carbon because carbon just doesn’t have enough electrons. It only has 4 total electrons within our second shell. And then here we have our little, meme in terms of remember we had Schrödinger’s cat. And remember with Schrödinger's cat, it was the possibility of being both alive and dead. So quantum mechanics states that I am simultaneously half alive, half dead. So this kind of goes in relationship to electrons where the electron could be in this position or it might not be. So, there's a half chance of it being there or not. So this kind of like goes hand in hand with that concept. So just remember we can't know both the speed and location of an electron, but we could talk about the probability, the most likely location of an electron. And that most likely location of an electron can be defined as orbitals.
Wave Function - Online Tutor, Practice Problems & Exam Prep
 Created using AI
Created using AIQuantum mechanics reveals that electrons exhibit both particle and wave characteristics, complicating their position and speed determination. The Heisenberg uncertainty principle states we cannot know both simultaneously, leading to a focus on probability through wave functions, denoted as Ψ. The probability of finding an electron is derived by squaring Ψ, resulting in atomic orbitals. Constructive and destructive interference of wave functions leads to bonding and antibonding molecular orbitals, respectively, influencing electron probability and chemical bonding.
This section deals with the basics of quantum mechanics. Don't worry too much about it, but this is some good general information.
Definition of an Atomic Orbital:

These funny orbital shapes represent the 3-D plots of the equations that describe the probability of finding electrons at any given place as their energy states increase.
The probability of finding electrons in a given place.
Video transcript
Atomic Orbital Interference
Instead of colliding into each other, wave functions have the ability to interfere with each other upon meeting.
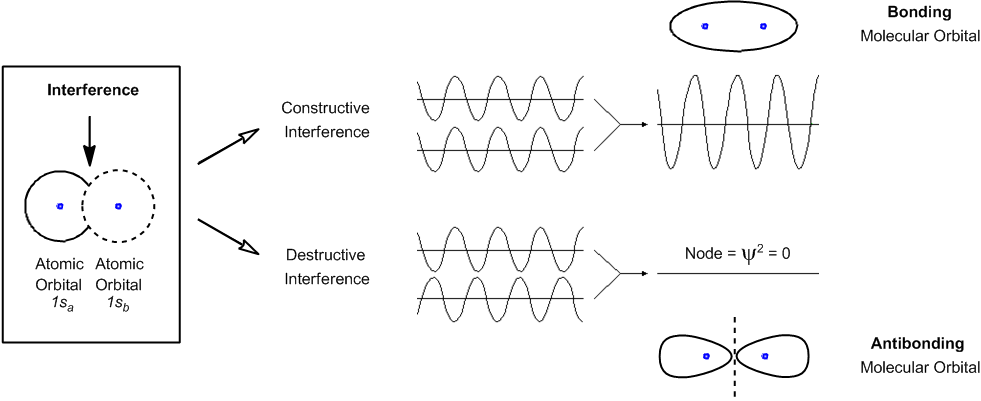
The type of interference determines if a new bond will be created between the two orbitals.
Constructive vs. destructive interference.
Video transcript
Let's keep going. So now let's talk about one more thing, in terms of quantum mechanics. And that is what I was hinting at earlier with the fact that these particles don't just act as particles. They don't just bump into each other. They act more like waves and to a certain extent. So, as waves, they have the ability to interfere with each other. So it's right in that word, Interfere. And what that means is that instead of colliding, they may actually interfere constructively or destructively.
So what does that mean? Well, think about it like this. Instead of thinking that these two orbitals are like balls and, like, they will hit each other and bounce off of each other. No. That's not how they act at all. Instead, think of these orbitals as waves, as regions of waves. And once these orbitals overlap with each other, they can either amplify each other or they can basically cancel each other out. You could also think about it for example, like a pool of water and I make two different splashes in two different places. Some of the waves are going to come together and make bigger waves. Right? But also, some of the waves are going to come together and actually cancel each other out. Maybe you've noticed that there will be some regions that are really big waves and some regions that have not that many waves. Okay? That's the difference between constructive and destructive interference.
So when I have constructive interference, the waves come together and they make waves of greater amplitude. Alright? With destructive interference, that means that these orbitals come together, but they wind up cancelling each other out in some way and actually making no net wave at the end.
Now what do these waves represent? I know you're thinking where is Johnny going with this? Well, when you increase the amplitude of your wave, what you're doing is you're increasing the chances of finding an electron that's in a certain place. Okay? So notice that I have this region of overlap right here. Okay? That is between the two. When I have constructive interference, what that means is that now the chances of finding electrons in this space right here are higher. Okay? When I have destructive interference, then what that means is that the chances of finding electrons in the middle are lower. Okay? And that's what I've drawn here at the top and the bottom. When they constructively interfere, that's what we call a bonding molecular orbital. And that's the whole idea behind a chemical bond. A chemical bond isn't just some random stick floating out in space. What it is, is that there's a higher mathematical probability of finding electrons in that region right here. And that's what we call a bond. So anytime you think of a bond now, think that's where electrons are likely to be. Okay?
Now the opposite is true when they destructively interfere. When they destructively interfere, that's called an anti-bonding orbital. What happens is that the chances of finding an electron in the middle actually go straight to zero. Remember that I said that Psi Square is the orbital or the relative probability. So, basically what we're saying is that there's no relative probability of finding electrons right in between there. By the way, just so you guys know, any area that has a probability of zero is called a node. That's just something random that you might need to know, just some terminology that you guys need to understand.
Do you want more practice?
More setsHere’s what students ask on this topic:
What is the Heisenberg Uncertainty Principle and how does it relate to wave functions?
The Heisenberg Uncertainty Principle states that it is impossible to simultaneously know both the exact position and exact momentum of an electron. This principle is crucial in quantum mechanics because it highlights the limitations of measuring subatomic particles. Wave functions, denoted as Ψ, are mathematical descriptions that provide the probability of finding an electron in a particular location. By squaring the wave function (Ψ²), we obtain the probability density, which helps us understand where an electron is likely to be found. This probabilistic approach is necessary due to the inherent uncertainties described by the Heisenberg Uncertainty Principle.
Created using AIHow do wave functions describe the behavior of electrons in atoms?
Wave functions (Ψ) describe the behavior of electrons in atoms by providing a mathematical representation of the electron's quantum state. These functions are solutions to the Schrödinger equation and correspond to specific energy levels of electrons. When we square the wave function (Ψ²), we obtain the probability density, which indicates the likelihood of finding an electron in a particular region around the nucleus. This probabilistic approach helps us understand the distribution of electrons in atomic orbitals, such as s, p, d, and f orbitals, each with distinct shapes and energy levels.
Created using AIWhat is the significance of constructive and destructive interference in wave functions?
Constructive and destructive interference in wave functions are significant because they influence the formation of molecular orbitals and chemical bonds. Constructive interference occurs when wave functions overlap and amplify each other, leading to a higher probability of finding electrons in the overlapping region. This results in bonding molecular orbitals, which stabilize the molecule. Destructive interference happens when wave functions overlap and cancel each other out, reducing the probability of finding electrons in the overlapping region. This creates antibonding molecular orbitals, which destabilize the molecule. Understanding these interferences helps explain the nature of chemical bonds and molecular stability.
Created using AIWhat are atomic orbitals and how are they related to wave functions?
Atomic orbitals are regions around an atom's nucleus where there is a high probability of finding an electron. They are derived from wave functions (Ψ), which are solutions to the Schrödinger equation for electrons in atoms. By squaring the wave function (Ψ²), we obtain the probability density, which describes the likelihood of finding an electron in a specific region. Atomic orbitals come in different shapes and energy levels, such as s (spherical), p (dumbbell-shaped), d, and f orbitals. These orbitals help us understand the electron configuration and chemical behavior of atoms.
Created using AIHow does the concept of nodes relate to wave functions and electron probability?
Nodes are regions in an atomic or molecular orbital where the probability of finding an electron is zero. They are directly related to wave functions (Ψ) because nodes occur where the wave function changes sign, resulting in Ψ² being zero. In atomic orbitals, nodes can be radial (spherical) or angular (planar). The presence and number of nodes help determine the shape and energy of the orbital. Understanding nodes is crucial for visualizing electron distribution and predicting chemical reactivity, as they indicate areas where electrons are unlikely to be found.
Created using AI
